
Wie äussert sich Cystische Fibrose? – Symptome
Symptome kurz nach der Geburt
Die Erbkrankheit Cystische Fibrose (CF) betrifft mehrere Organe. Deshalb treten verschiedene Symptome auf, einige oft schon kurz nach der Geburt:
- Das Mekonium oder «Kindspech» geht nicht ab: Es kommt zu einem Darmverschluss schon vor der ersten Stuhlentleerung.
- Die Kinder haben einen «salzigen Geschmack» auf der Haut.
- Die Babys bleiben untergewichtig, leiden unter Blähungen und Bauchschmerzen, ihr Stuhl ist gräulich und fettig.
- Schon früh leiden die Kleinen unter chronischem Husten und Atembeschwerden.
Zäher Schleim in den Atemwegen
Bei Menschen mit Cystischer Fibrose produzieren die schleimbildenden Zellen ein sehr zähes Sekret. In den Atemwegen behindert dieser zähe Schleim den Reinigungsmechanismus; eingeatmete Schmutzteilchen und Krankheitskeime bleiben hängen. Die Folgen sind wiederkehrende Atemwegsinfektionen und chronischer Husten. Bei fortgeschrittener Erkrankung wird das Lungengewebe zerstört, was zu Atemnot und Sauerstoffmangel führt.
Verdauung ist eingeschränkt
Bei Menschen mit Cystischer Fibrose verstopft zäher Schleim die Gänge der Bauchspeicheldrüse. Verdauungsenzyme können nicht in den Darm gelangen und die Nahrung wird ungenügend verdaut. Deshalb gedeihen betroffene Kinder oft schlecht, Erwachsene sind untergewichtig.
Weitere Symptome und Begleiterkrankungen bei Erwachsenen sind: Entzündung der Leber und Gallenblase, Diabetes und Knochenschwund (Osteoporose).
Ursachen & Behandlungsmöglichkeiten
Weshalb tritt Cystische Fibrose auf? – Ursachen
Die Cystische Fibrose entsteht durch einen Fehler im Erbgut, den die Eltern an die Kinder weitergeben. Den genauen Ort dieses Fehlers fanden Wissenschaftler im Jahr 1989: Auf Nummer 7 der insgesamt 23 Chromosomenpaare ist bei Menschen mit Cystischer Fibrose eine falsche Information gespeichert. Diese verhindert, dass ein wichtiges Eiweiss, das CFTR-Protein (Cystic Fibrosis Transmembrane Conductance Regulator), richtig hergestellt wird. Dieses sorgt bei gesunden Menschen dafür, dass Schleim und Sekrete ausreichend flüssig sind. Ist es defekt, sind Wasser- und Salzhaushalt im Körper gestört: Die Körperflüssigkeiten sind zähl und klebrig und verstopfen lebenswichtige Organe wie die Bauchspeicheldrüse, den Darm und vor allem die Lunge.
Gesunde Eltern, kranke Kinder
Etwa jeder zwanzigste Mensch in der Schweiz trägt das defekte Gen auf dem Chromosom Nummer 7. Dennoch erkrankt eines von 3300 Neugeborenen an Cystischer Fibrose. Warum? Damit ein Kind an Cystischer Fibrose erkrankt, müssen beide Eltern den Fehler in ihren Genen tragen und beide Eltern müssen das fehlerhafte Gen weitergeben. Erhält ein Kind nur von einem Elternteil ein defektes Gen, ist es zwar Merkmalsträger, aber gesund
Cystische Fibrose nachweisen – Diagnose
Neugeborenen-Screening
Seit 2011 werden alle Neugeborenen auf Cystische Fibrose getestet. Am vierten Tag nach der Geburt entnehmen Fachpersonen dem Kind einige Tropfen Blut aus der Ferse. Dieses wird auf zehn Hormon- und Stoffwechselkrankheiten untersucht, unter anderem auf Cystische Fibrose.
Schweisstest
Ergibt das Neugeborenen-Screening ein auffälliges Resultat, kann ein Schweisstest die Diagnose bestätigen. Bei Säuglingen mit Cystischer Fibrose ist der Salzgehalt im Schweiss erhöht. Bei 98 Prozent der Betroffenen ist der schmerzfreie Test positiv.
Gentest
Bei einem Gentest werden ein Tropfen Blut oder ein Abstrich der Mundschleimhaut auf den Gendefekt untersucht.
Diagnose vor der Geburt
Bereits in der frühen Schwangerschaft lässt sich feststellen, ob das Ungeborene an Cystischer Fibrose erkrankt ist oder nicht. Dazu wird zwischen der achten und der zwölften Schwangerschaftswoche eine Gewebeprobe aus dem Mutterkuchen entnommen (Chorionzottenbiopsie) oder zwischen der 14. und 16. Schwangerschaftswoche eine Fruchtwasserpunktion (Amniozentese) durchgeführt.
Höhere Lebenserwartung – Behandlung
Sämtliche Behandlungsformen zielen darauf ab, die Funktion der betroffenen Organe möglichst lange zu erhalten. Dank neuer Möglichkeiten sind die Lebenserwartung und die Lebensqualität der Patientinnen und Patienten heute deutlich höher als früher. Neugeborene, bei denen die Krankheit beim Neugeborenen-Screening entdeckt wird, haben gute Chancen, das Rentenalter zu erreichen.
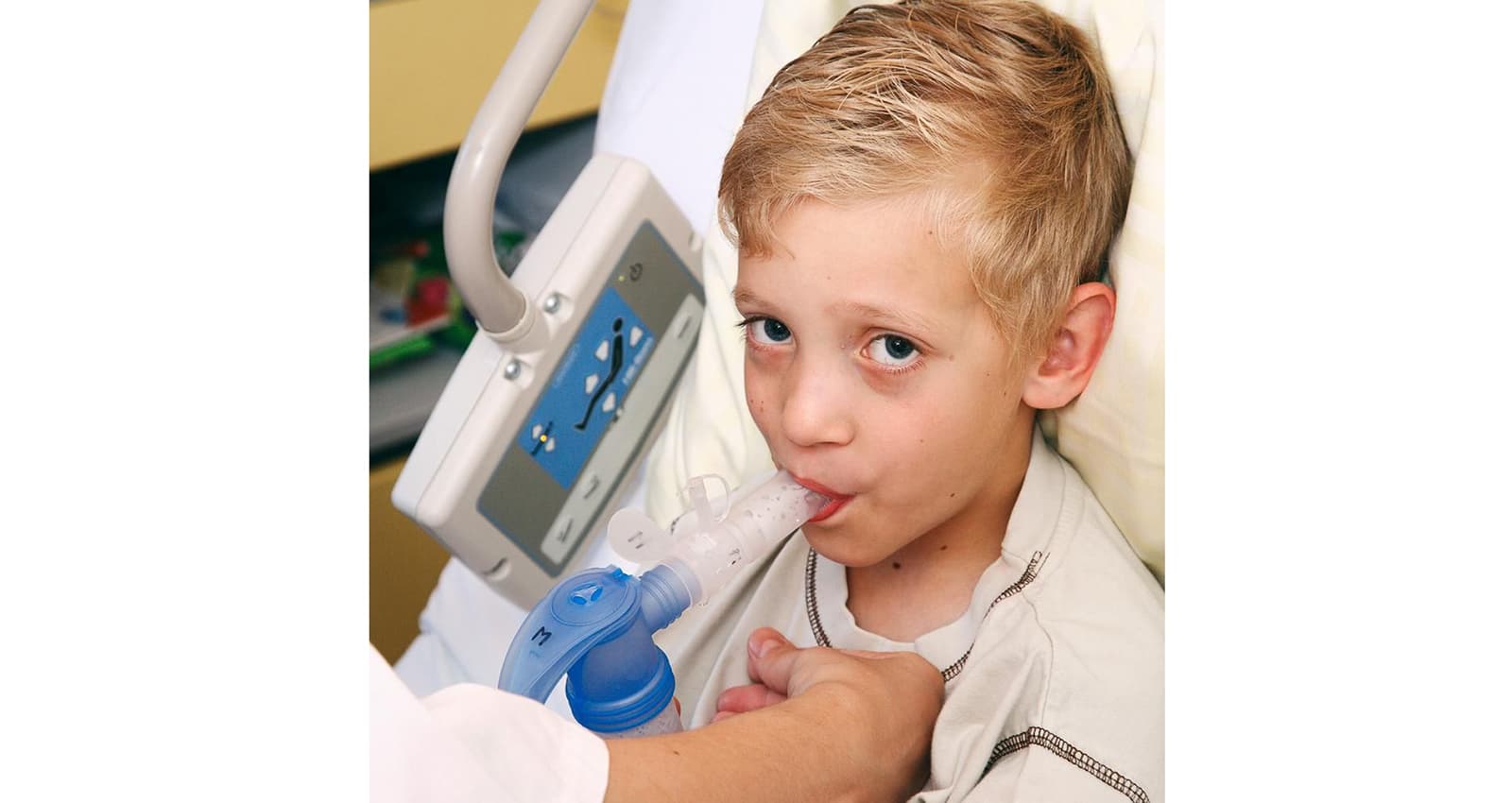
Atemphysiotherapie verbessert Lungenfunktion
Physiotherapeutische Behandlungsmethoden begleiten die Betroffenen ihr Leben lang. Zum Angebot physiotherapeutischer Betreuung gehören in erster Linie Inhalations- und Atemphysiotherapie, daneben aber auch Übungen für Kraft und Ausdauer sowie Entspannungstechniken. Bei der Atemphysiotherapie können Betroffene mit speziellen Übungen ihre Lungenfunktion verbessern sowie den zähen Schleim in den Bronchien lösen und nach aussen befördern. Dadurch fällt das Atmen leichter und Krankheitserreger nisten sich nicht so schnell ein.
Antibiotika gegen bakterielle Infektionen
Die häufigen Infektionen der Atemwege tragen entscheidend zur Zerstörung der Lungen von Betroffenen bei. Verantwortlich dafür sind oft Bakterienarten, die sich mit Antibiotika unter Kontrolle halten lassen. Bei häufigem Gebrauch von Antibiotika können jedoch als mögliche Nebenwirkungen Allergien oder Resistenzen auftreten.
Chloridkanal wieder funktionsfähig machen
Mit neuen Medikamenten, sogenannten CFTR-Modulatoren (Cystic Fibrosis
Transmembrane Conductance Regulator) ist es seit Kurzem möglich, die Funktion des CFTR-Proteins (Chloridkanal) zu verbessern oder wieder herzustellen. Damit können viele symptomatische Therapien reduziert und teilweise ganz gestoppt werden.
Mehr Kalorien und Verdauungsenzyme
Wegen den häufigen Infektionen und der gestörten Funktion der Bauchspeicheldrüse leiden die Betroffenen häufig an Mangelernährung. Deshalb brauchen sie mehr Kalorien und Vitamine als gesunde Menschen. Damit der Körper die Nährstoffe aufnehmen kann, müssen Menschen mit Cystischer Fibrose vor und während den Mahlzeiten Verdauungsenzyme einnehmen.
Lungentransplantation
Ist die Zerstörung der Lunge trotz intensiver Therapie weit fortgeschritten, ist die Lungentransplantation eine Therapiemöglichkeit. Diese Massnahme ist angezeigt, wenn die Lebenserwartung auf weniger als zwei Jahre geschätzt wird, eine stark eingeschränkte Lungenfunktion vorliegt und häufig schwere Infektionen auftreten. Der Entscheid für oder gegen eine Transplantation hängt von vielen verschiedenen Faktoren ab.
Kommt es bei Cystischer Fibrose zu einer Transplantation, werden beide Lungenflügel transplantiert. Hauptsächliche Probleme nach einem solchen Eingriff sind Infektionen und die Abstossung des neuen Organs.

Kurse & Angebote für Betroffene
Haben Sie Fragen? Wir helfen weiter!
Die kantonalen Lungenligen leisten umfassende Beratung und Betreuung. Dies mit dem Ziel, dass Betroffene möglichst beschwerdefrei leben können. Die kantonale Lungenliga ihn Ihrer Nähe hilft Ihnen bei Ihren Fragen gerne weiter.
